



























MASLD, MASH, and Fibrosis Research Reagents
Metabolic dysfunction-associated steatohepatitis (MASH; formerly non-alcoholic steatohepatitis, NASH), metabolic dysfunction-associated steatotic liver disease (MASLD; formerly non-alcoholic fatty liver disease, NAFLD), and fibrosis are a set of liver pathologies that have increased in prevalence over recent decades and are now on the verge of reaching global epidemic proportions. These conditions develop from the accumulation of fat in the liver— the first and reversible event in the physio-pathological progression from a healthy liver to MASH. This abnormal and significant lipid accumulation in the liver, referred to as hepatic steatosis, simple steatosis (SS), or non-alcoholic fatty liver (NAFL), results from a substantial increase in the flux of free fatty acids into the organ. This condition is thought to arise from dysregulation of key aspects of lipid metabolism, including lipogenesis, beta-oxidation, and the export/uptake/storage axis.
Revvity has developed a panel of ready-to-use assays to monitor biomarkers, phosphoproteins, and transcription factors involved in the development and progression of MASH, MASLD, and fibrosis. In addition to our commitment to providing the best possible tools for your research, we also offer comprehensive resources on MASH, including guides, infographics, application notes, tips, and data.

Learn more about the pathogenesis and key signaling pathways involved in MASH and MASLD in our dedicated guide.
Take a quick look at the state of the growing MASH and MASLD epidemics and the associated risk factors in this infographic.
For research use only. Not for use in diagnostic procedures.

Hepatic steatosis
The accumulation of fat in the liver is the first and main reversible event to occur in the physio-pathological progression from a healthy liver to MASH. This abnormal and significant lipid accumulation, referred to as hepatic steatosis or simple steatosis (SS), results from a steep increase of free fatty acid deposition within liver tissue. This condition is thought to arise from dysregulation of key aspects of lipid metabolism, including lipogenesis, beta-oxidation mechanisms, and export/uptake/storage of lipids. As such, it is often associated with obesity, with which it shares several metabolic causes, including insulin resistance and lipid accumulation in organs.
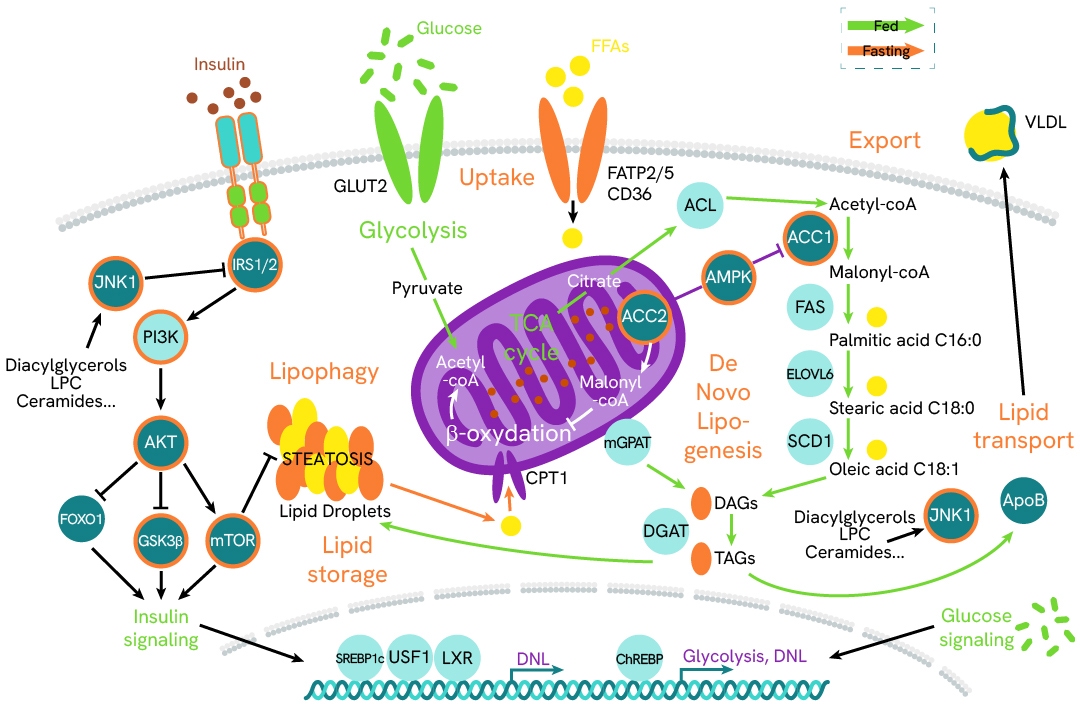
In hepatocytes, these mechanisms are regulated by two key pathways: insulin signaling through its receptor and downstream phosphorylation cascade, and the conversion of acetyl-CoA into substrates for diglyceride synthesis. Revvity has developed a panel of ready-to-use assays to monitor the key biomarkers, phosphoproteins, and transcription factors involved in these signaling cascades.
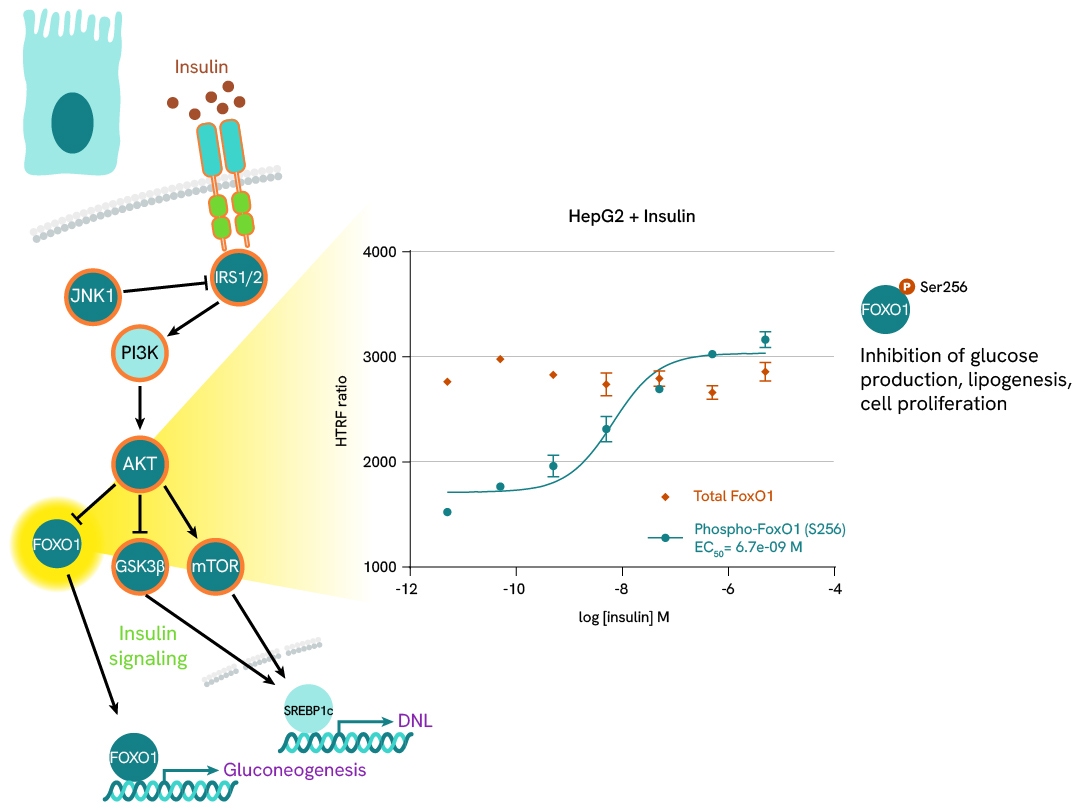
FOXO1 phosphorylation as a marker of insulin signaling
Insulin stimulation of hepatocytes activates the PI3K-AKT pathway, leading to the phosphorylation of FOXO1 at serine 256 and its subsequent translocation to the nucleus. In the context of insulin resistance and obesity, insulin levels rise to maintain normal glucose tolerance. This can be mimicked in cell models by treating with high concentrations of insulin.
In this case study, HepG2 cells were plated at 25,000 cells/well and stimulated with increasing concentrations of insulin for 1 hour at 37°C. After a 30-minute lysis incubation time, total and phospho-FOXO1 levels were measured, showing an increase in phosphorylation with increasing doses of insulin, while total protein levels remained stable.
Learn more about this experiment and other studies on MASH pathways in the corresponding application note.
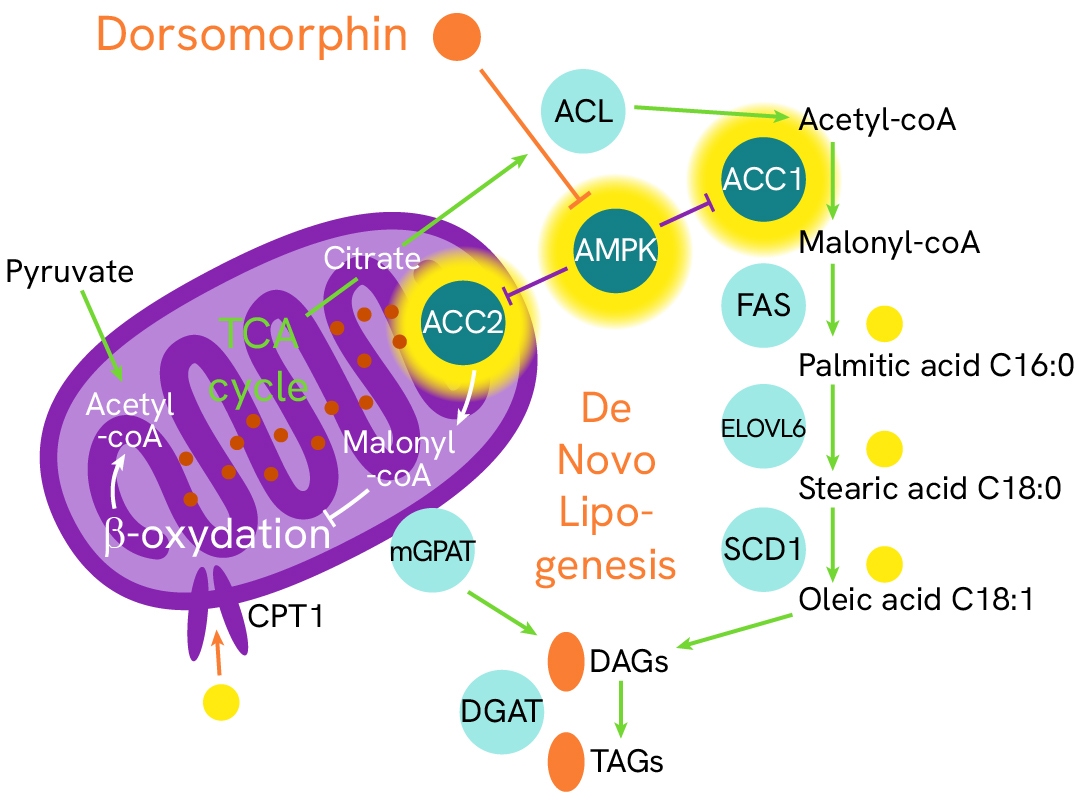
ACC2/AMPK/ACC1 signaling regulates de novo lipogenesis in hepatic steatosis onset
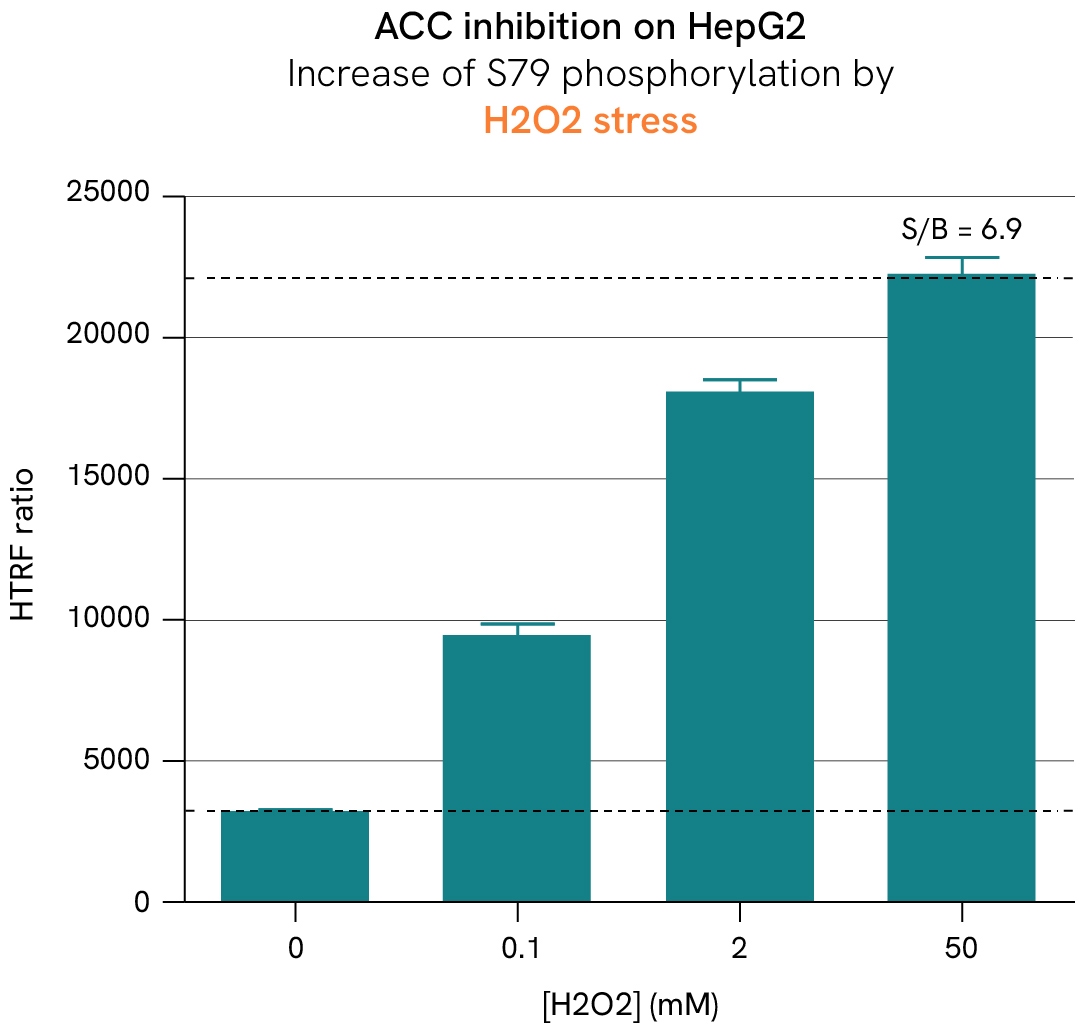
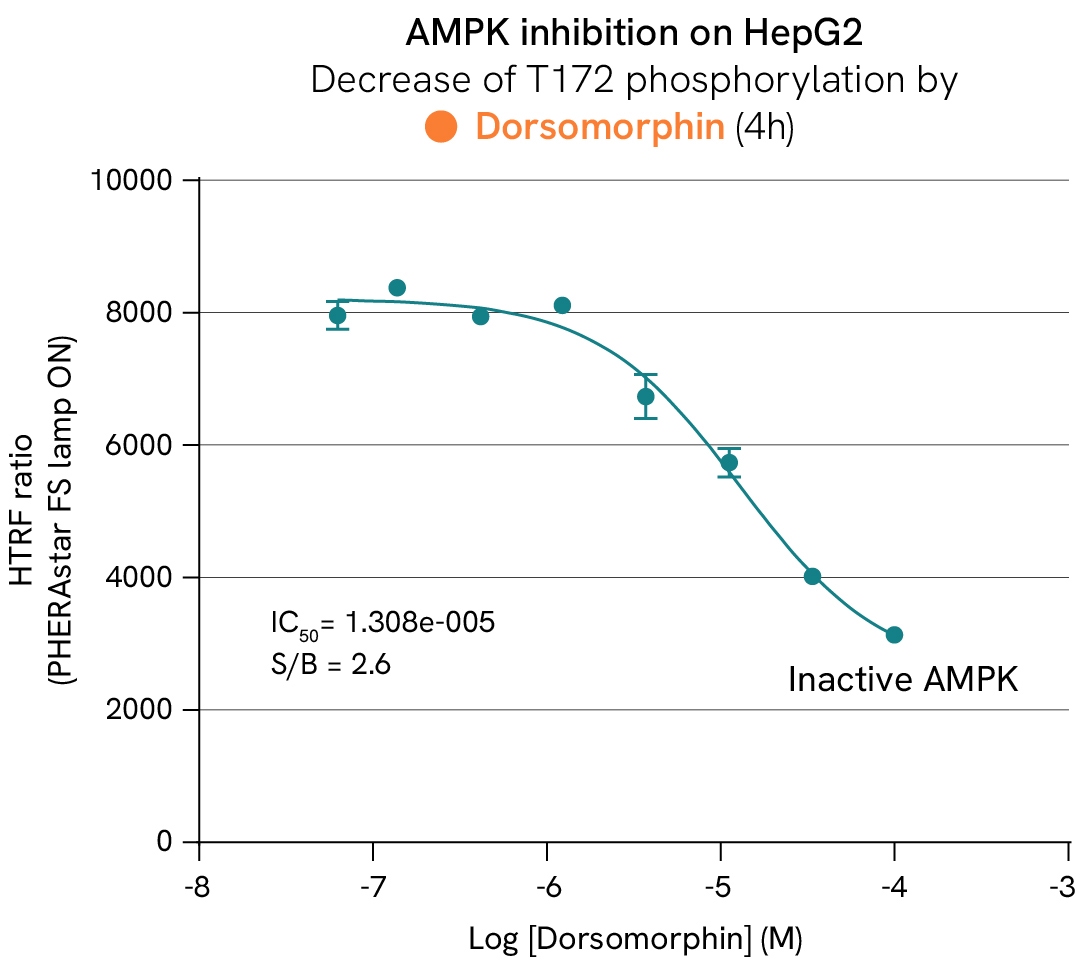
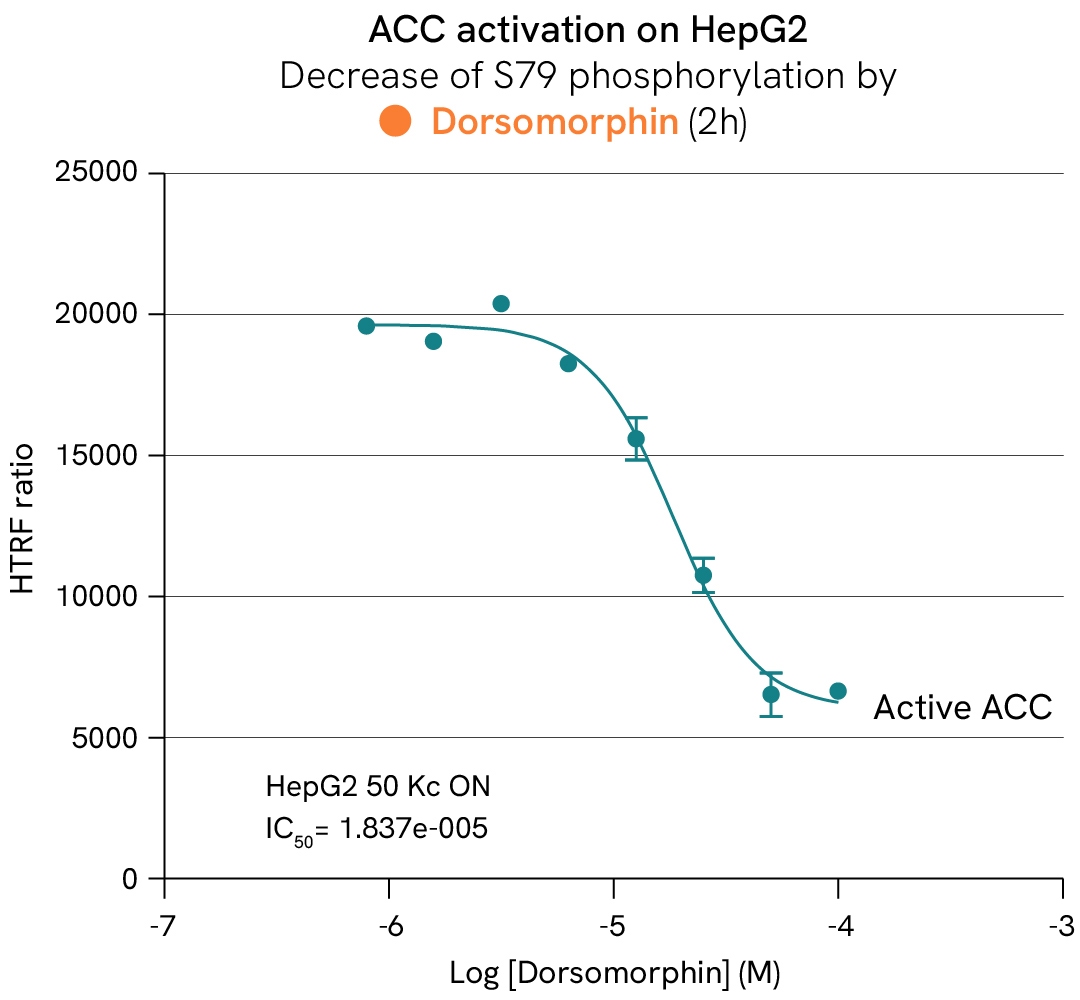
AMP-activated protein kinase (AMPK) and its ACC substrates function as metabolic sensors and switches that regulate the fate of acetyl-CoA based on the availability of ATP in cells. In the absence of ATP, AMPK is activated by phosphorylation and subsequently phosphorylates ACC1/2 at Ser79, inhibiting their activity. This suppression inhibits the conversion of acetyl-CoA into malonyl-CoA—a key substrate for fatty acid biosynthesis and a repressor of lipid beta-oxidation—thereby reducing lipid droplet accumulation in cells. Conversely, inhibiting AMPK promotes ACC activity and restores a healthy balance between beta-oxidation and lipogenesis.
In this case study, HepG2 cells were stimulated with increasing concentrations of dorsomorphin at 37°C or subjected to H2O2 stress. After lysis, the phosphorylation levels of AMPK, ACC1, and ACC2 were measured.
Progression of MASH/MASLD
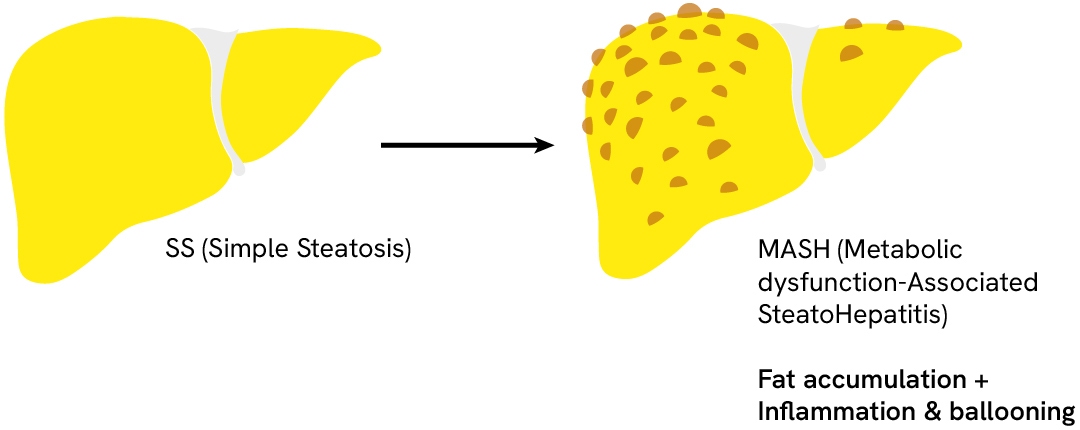
The pathogenic mechanisms of MASH are complex and multifactorial. Recent findings have challenged prior knowledge of the pathology and proposed a pathogenesis that involves organ-organ interactions and multiple parallel processes. These include, but are not limited to, genetic predisposition, insulin resistance, adipose tissue dysfunction, abnormal lipid metabolism, lipotoxicity, altered production of inflammatory mediators, and dysregulation of the gut-liver axis and innate immunity.
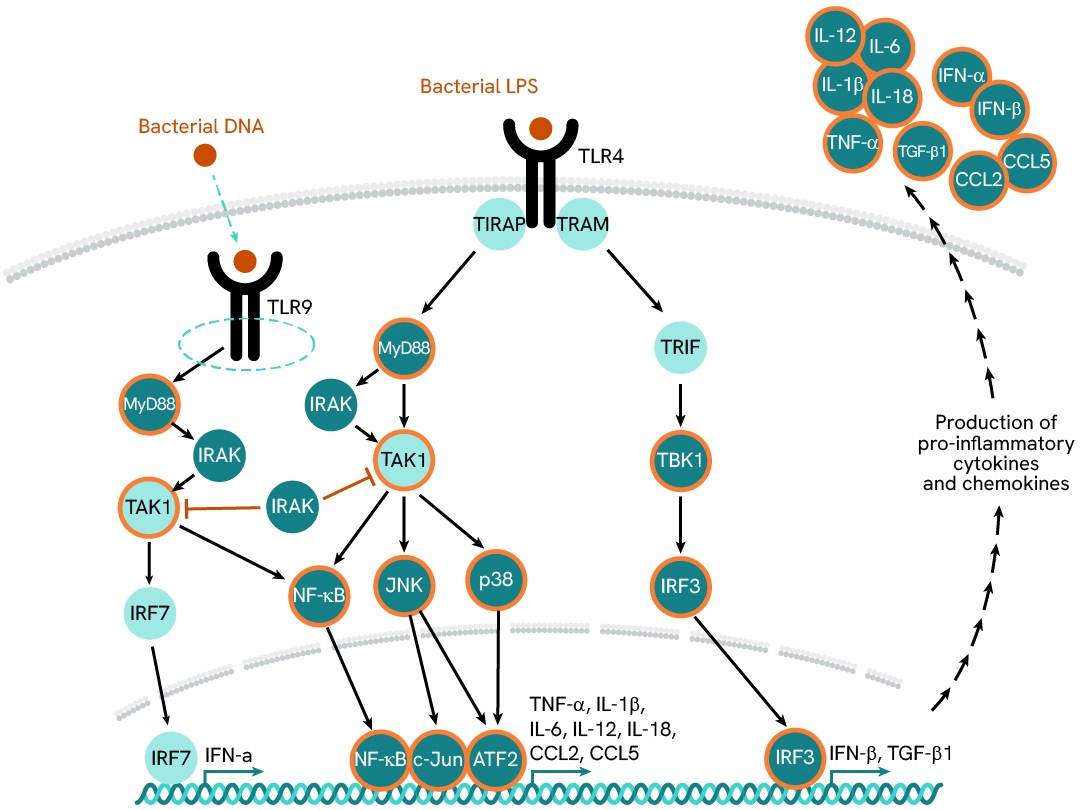
Investigation of LPS/TLR4-driven activation of Kupffer cells
In MASH patients, the liver is continuously exposed to an overload of gut-derived bacterial products, which activate liver-resident macrophages— known as Kupffer cells—via their TLRs. These innate immune cells signal primarily through two TLR-associated pathways: MyD88 and TRIF/TBK1.The MyD88 pathway promotes the secretion of pro-inflammatory mediators (such as CCL2, IL6, IL12, and IL1β), while the TRIF/TBK1 pathway drives the expression of pro-fibrogenic TGF-β1. Chronic activation of these cells results in a pro-inflammatory imbalance and a feedback loop, which is detrimental to liver function. Simultaneously, the release of pro-fibrotic mediators contributes to a gradual increase in tissue stiffness and progressive loss of function.
In this case study, we explore the effects of LPS stimulation on the expression of a panel of cytokines (TGFβ1, TNFα, IL6, and CCL2) in supernatants, as well as the activation of downstream signaling phosphoproteins (phospho-p38 and phospho/total-TBK1) in cell lysates. Assays were run in ImKC mouse Kupffer cells plated at 100,000 cells/well and stimulated with varying concentrations of LPS.
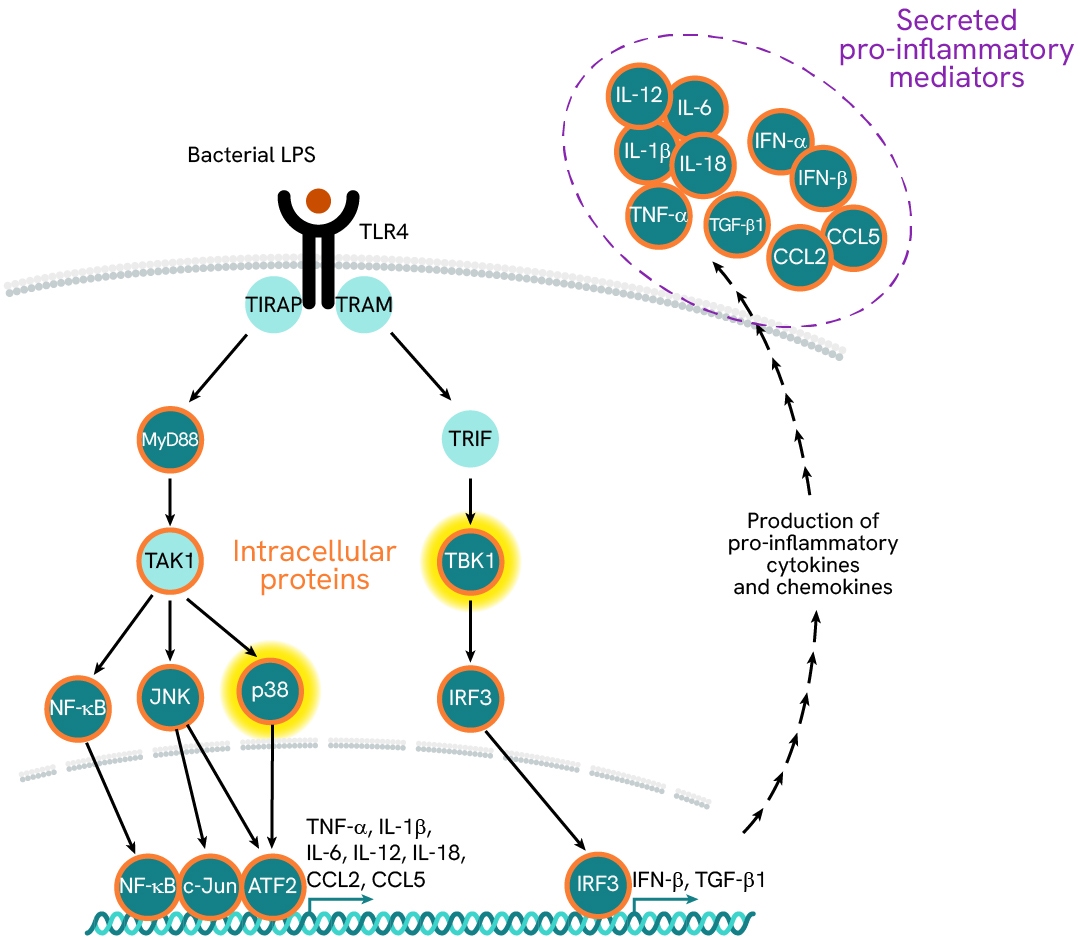
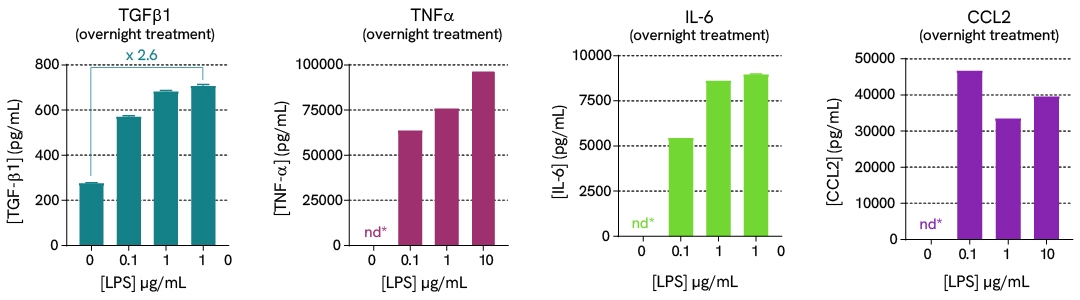
Learn more about this experiment and other investigations on MASH pathways in the corresponding application note.
Liver fibrosis
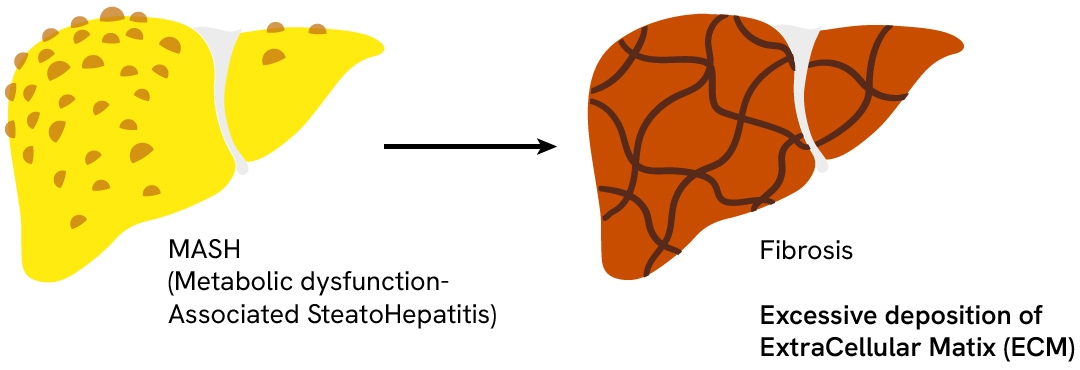
Hepatic fibrosis represents a late stage in MASH progression where the overactivation of matrix-depositing cells is a healing response to chronic liver injury and inflammation. Fibrosis is characterized by excessive deposition of extracellular matrix (ECM) due to an imbalance between ECM synthesis and degeneration. In MASH, damaged hepatocytes express inflammatory mediators that recruit and activate immune cells. The lasting inflammation of local macrophages (Kupffer cells) promotes the expression of TGF-β1— a key pro-fibrogenic mediator that triggers quiescent hematopoietic stem cell (HSC) differentiation into myofibroblasts. These activated myofibroblasts are characterized by the presence of α-SMA-inclusive actin fibers, which confer contractile properties. They serve as the primary matrix-secreting cells and drive liver fibrosis by depositing abnormally high amounts of extracellular matrix components—mostly collagens—in the liver tissues.
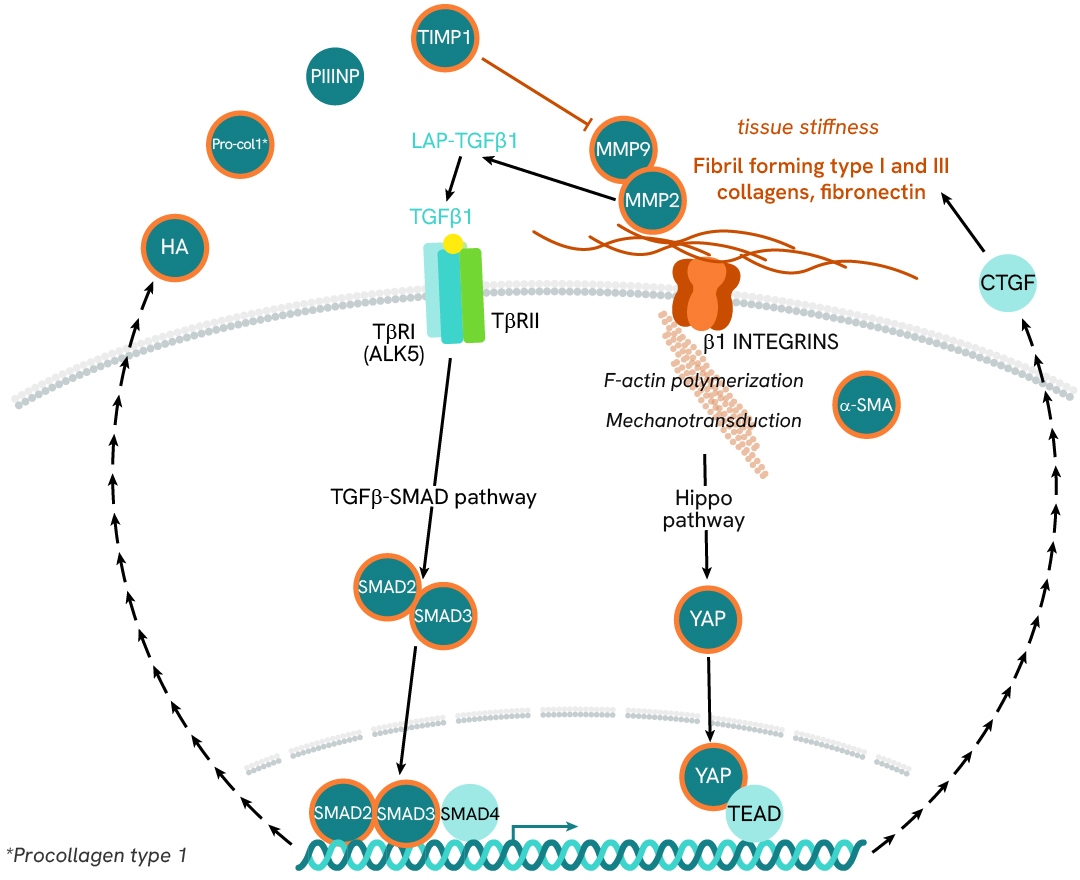
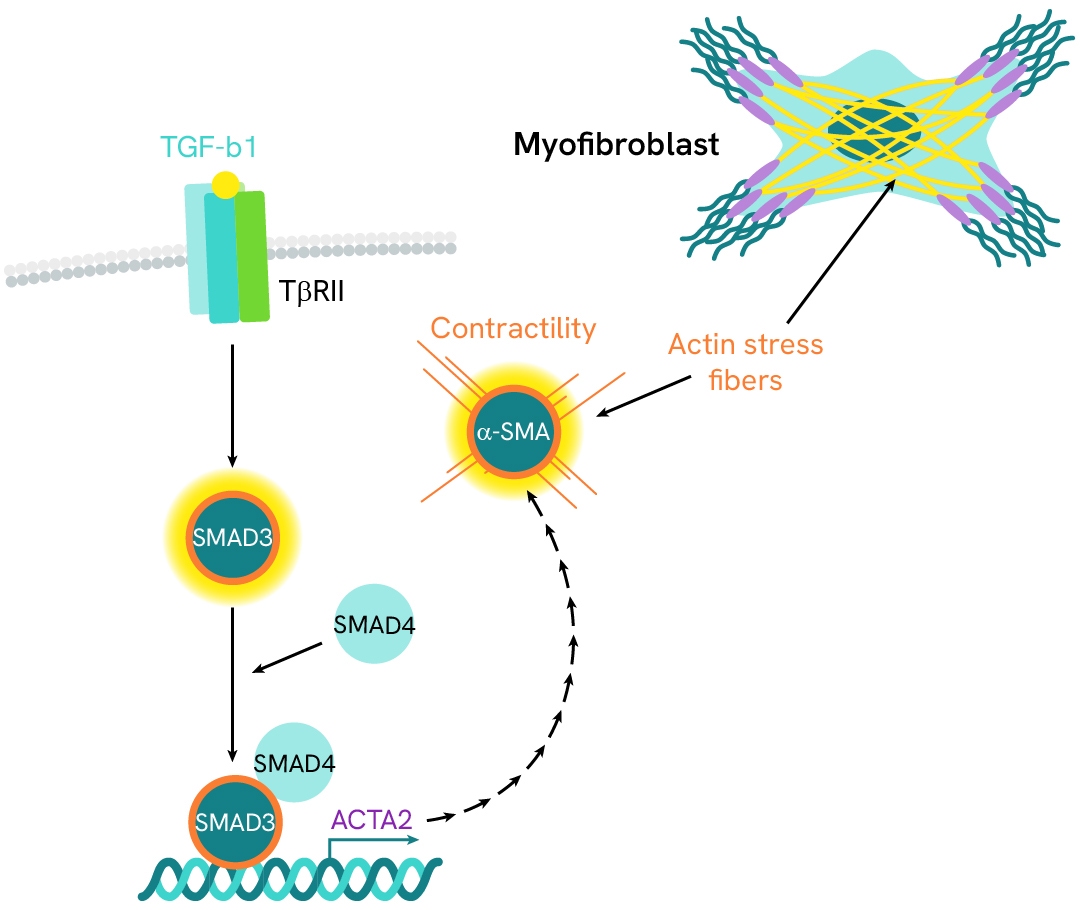
Investigation of HSC activation and differentiation with SMAD signaling and α-SMA expression
α-smooth muscle actin (α-SMA) is an actin protein with high homology to the five other actin isoforms. Its expression is primarily induced by the pro-fibrogenic cytokine TGF-β1, which signals through the SMAD pathway. α-SMA is a key marker of hepatic stellate cell (HSC) transition into myofibroblasts and of MASH progression toward fibrosis. It contributes to the formation of stress fibers that confer the contractile properties characteristic of activated myofibroblasts.
In this case study, we study the differentiation of HSC into myofibroblasts by monitoring α-SMA expression and SMAD3 phosphorylation in LX2 HSC cells, in presence of either TGF-β1 or TGF-β1 receptor subunit ALK5 inhibitor.
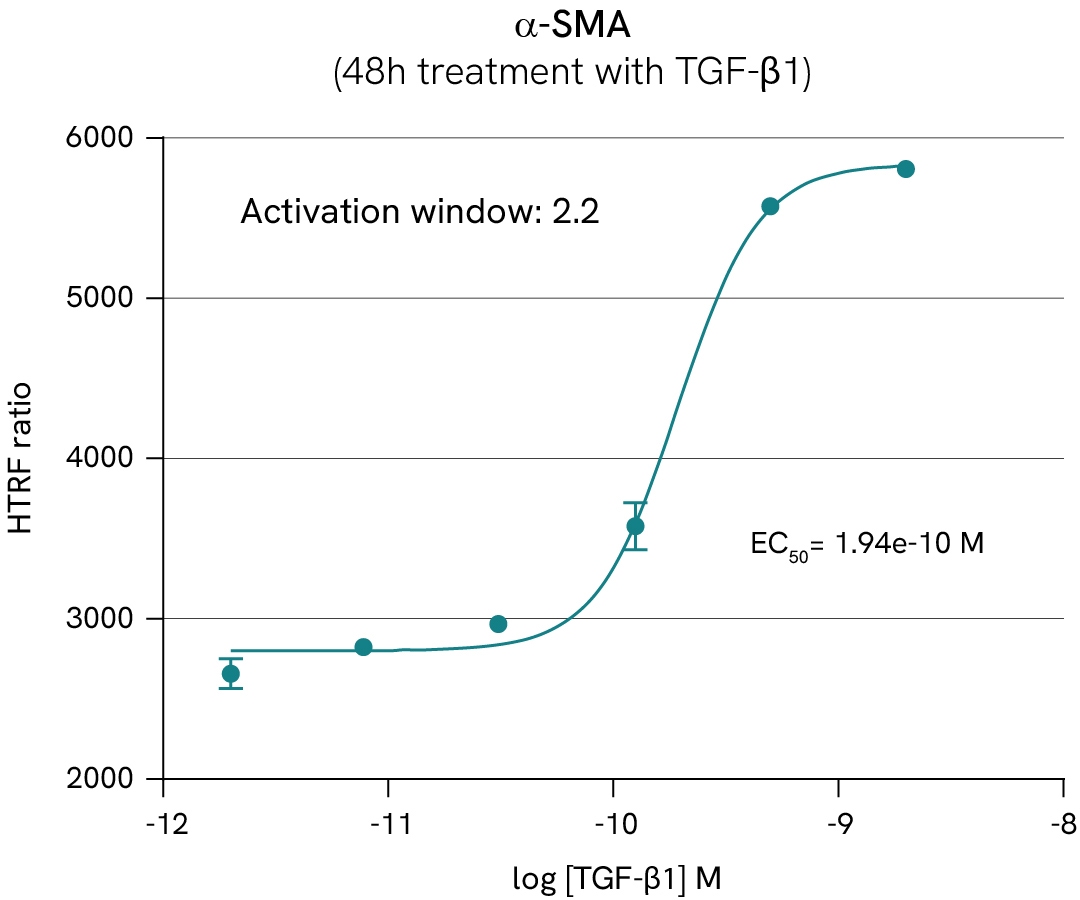
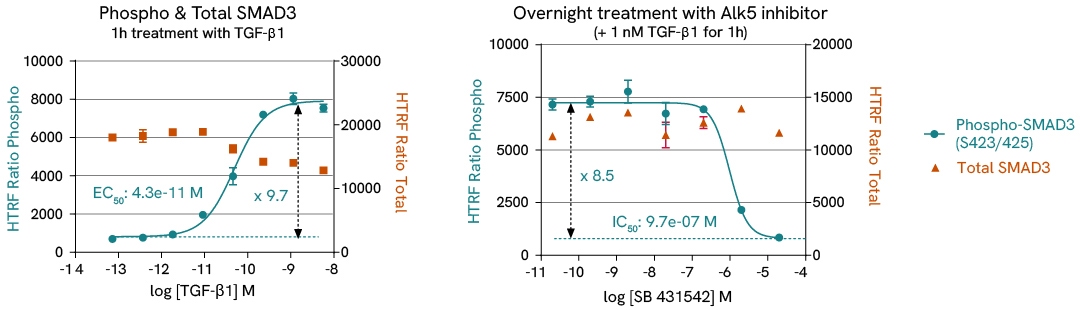
Learn more about this experiment and other investigations on liver fibrosis pathways in the corresponding application note.
Featured resources

