





























The NovaSeq® X/X Plus sequencers1 are llumina’s® highest throughput sequencing platforms, delivering up to ~ 26 billion reads per run and achieving ≥ 80% of bases at Q40 in optimal runs2. These instruments are engineered for applications demanding immense scale, such as population genomics, ultra-deep whole-genome sequencing, and high-throughput multiomic studies.
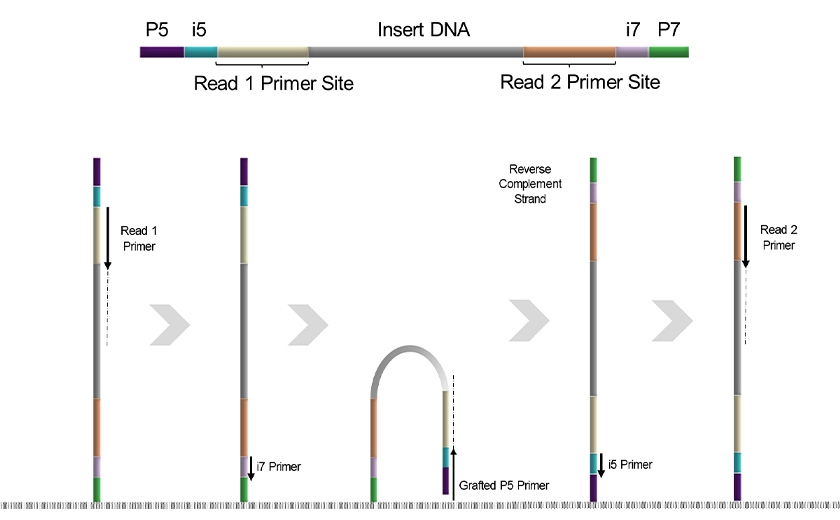
Dual-index sequencing on a paired-end flow cell follows one of two workflows, depending on the Illumina system. The Illumina® NovaSeq® X/X Plus sequencers, like most of Illumina® instruments, use the reverse complement workflow, also known as “workflow A”. In this workflow Read 1 and Index 1 (i7) are synthesized from the P7-anchored strand, generating the forward sequence of the insert. Afterward, the original template is denatured and then bridge-amplified to produce a full reverse complement strand anchored at the P5 adapter. Index 2 (i5) and Read 2 are then generated from this newly synthesized strand3 (Figure 1).
Figure 1. Reverse complement workflow used by Illumina® NovaSeq® X/X Plus sequencers for dual-index sequencing.
This reverse-strand synthesis step can lower Read 2 quality compared to Read 1 due to several factors, that are listed in Table 1.
| Issue | How it impacts Read 2 |
|---|---|
| Polymerase error in new strand synthesis | If the polymerase misincorporates a base when creating the reverse complement, all molecules in that cluster carry that error, resulting in systematic rather than random base-calling errors4. |
| Incomplete strand synthesis | Incomplete synthesis can leave a portion of clusters without a full template at the start of Read 2, reducing per-cycle intensity and increasing the risk of phasing/prephasing errors in the early cycles. |
| Template loss | Clusters can lose templates during the additional washes and denaturation steps, reducing signal intensity of Read 2. Loss of diversity due to cluster dropout can also cause issues in registration and color normalization. |
| Accumulated phasing/pre-phasing |
Even with optimized XLEAP-SBS chemistry, phasing/pre-phasing rates are not zero. Because clusters are already a full read’s worth of cycles into the run by the time Read 2 starts, fewer clusters remain perfectly synchronized. This means that in a given cycle, the cluster’s fluorescent signal is now a mixture of bases instead of a clean, single-base call. |
In most high-quality libraries, Read 2 Q scores are 1–3 points lower than Read 15. The gap widens for runs with long reads, because phasing/prephasing has more time to accumulate. In terms of error rate, this difference implies that Read 2 contain ~ 1.6X more errors than Read 1. The absolute error rate increase is still small (from 1 in 10,000 to ~ 1 in 6,300), but it becomes important in low-variant-frequency detection or when looking for rare molecules.
PCR-free workflows and Read 2 quality on Illumina® NovaSeq® X/X Plus sequencers
PCR-free library preparation removes amplification bias and can be performed with Y-shaped full-length adapters such as the NEXTFLEX™ Unique Dual Index Barcodes
 NEXTFLEX Unique dual index barcodes (10NT, 1-16)
. In some NovaSeq® X/X Plus runs, users have observed a significant drop in R2 quality in PCR-free workflows, However, adding a post-ligation double-SPRI cleanup (for example 0.5x + 0.8x) has been shown to restore the Q scores for Read 2 on when using NEXTFLEX Unique Dual Index Barcodes.
NEXTFLEX Unique dual index barcodes (10NT, 1-16)
. In some NovaSeq® X/X Plus runs, users have observed a significant drop in R2 quality in PCR-free workflows, However, adding a post-ligation double-SPRI cleanup (for example 0.5x + 0.8x) has been shown to restore the Q scores for Read 2 on when using NEXTFLEX Unique Dual Index Barcodes.
We recommend that users performing PCR-free workflows on NovaSeq® X/X Plus sequencers, with our barcodes or with other full-length adapters, adopt this double-SPRI cleanup as best practice step. Although the mechanism is not fully understood, our working hypothesis is that in PCR-free workflows, unligated barcodes remaining after ligation are more likely to persist into the final pool. Y-shaped adapters are denatured as part of the standard Illumina cluster generation process.
If these unligated species are present at high levels, they may anneal to the flow cell oligos, potentially blocking bridge amplification, or pair with another template strand to initiate cluster amplification. In the latter case, this could contribute to index hoping which has been reported by some users when using full-length barcodes on the Illumina® NovaSeq® X/X Plus Sequencers.
By performing the double-SPRI cleanup, the proportion of full-length, structurally intact molecules in the library increases. This, in turn, means that more clusters survive intact through to Read 2, improving the signal-to-noise ratio during the early Read 2 cycles. If this hypothesis is correct, lowering the adapter concentration during ligation should further reduce the amount of unligated product at the end of this step.
References:
- NovaSeq X Series | Production scale, ultra-high-throughput sequencers
- NovaSeq X v1.2 software enables sequencing with 80% of bases >= Q40
- Dual-Indexed Workflow on a Paired-End Flow Cell
- Quality Scores for Next Generation Sequencing | Illumina Knowledge
- Why has the reverse read 2 a worse quality than the forward read 1 in Illumina sequencing?



