





























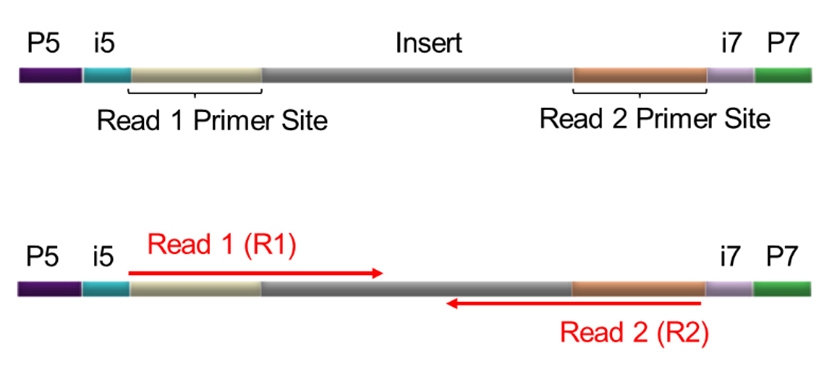
Illumina® paired-end sequencing provides sequence data from both ends of each DNA fragment (insert), generating two reads, Read 1 (R1) and Read 2 (R2). They are independent but linked by their physical origin. R1 corresponds to the sequence immediately downstream of the P5 adapter, while R2 corresponds to sequence that immediately upstream of the P7 adapter. (Figure 1).
In WGS
 NEXTFLEX Rapid XP V2 DNA-Seq Kit
, the availability of two reads per fragment improves the resolution of complex genomic regions, where repetitive elements often exceed single-read length. By providing an approximate insert size between reads, paired reads help disambiguate mapping in repeat or segmentally duplicated regions, improving single‑nucleotide and indel variant calling.
NEXTFLEX Rapid XP V2 DNA-Seq Kit
, the availability of two reads per fragment improves the resolution of complex genomic regions, where repetitive elements often exceed single-read length. By providing an approximate insert size between reads, paired reads help disambiguate mapping in repeat or segmentally duplicated regions, improving single‑nucleotide and indel variant calling.
Paired-end sequencing also enhances the detection of structural variants. Normally, R1 aligns to one strand of the reference genome, and R2 to the opposite strand, oriented inward (Figure 2). This pattern (often called forward–reverse although it does not imply that R1 is uniformly “forward”) is the hallmark of a correctly constructed library. Deviations from this pattern can signal library prep issues or genuine variants, such as chromosomal rearrangements in cancer.
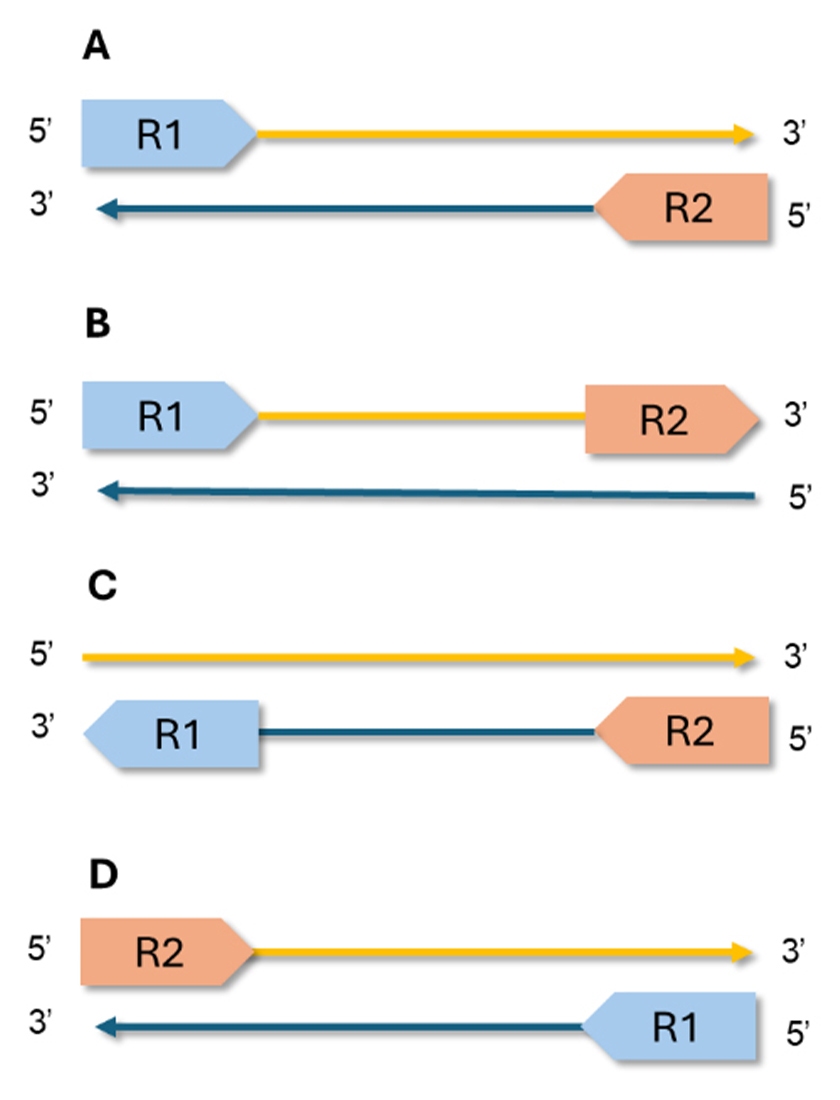
Figure 2. Common mapping orientations for paired-end reads in WGS. A) forward-reverse, expected; B and C) forward-forward, and reverse-reverse, same direction reads, typically artifacts or unusual library prep. D) reverse-forward, often seen in structural variants such as inversions.
Causes of the appearance of abnormal read pairs
In standard Illumina® whole genome sequencing (WGS), properly oriented forward‑reverse pairs typically comprise >95%-98% of mapped reads. Elevated levels of discordant orientations (forward‑forward, reverse‑reverse, reverse‑forward) can stem from both biological and technical factors.
Biological causes of abnormal read pairs
- Structural variants: Large-scale genomic changes such as deletions, insertions, inversions, translocations, and duplications often leave a signature of clustered discordant read pairs in defined regions. These patterns reflect true genomic alterations that distinguish the sample’s genome from the reference sequence..
Technical causes of abnormal read pairs
- Alignment errors: Low‑quality reads and repetitive sequences challenge aligners (e.g., BWA, Bowtie2), often yielding incorrect placements and discordant orientations. Some researchers have noted that in practice, abnormal pairs are more frequently false alignments rather than true structural variants, even in cancer genomes.
- Sequencing errors: Errors in the sequencing process can interfere with read alignment and result in discordant reads.
- Insert size variability: A broad insert size distribution, or fragments shorter than twice the read length (leading to overlap and adapter read‑through), can produce clipped or misoriented alignments.
- Chimeric inserts: Randomly ligated fragments or FFPE‑induced chimeras (e.g., due to microhomology during end‑repair) generate false discordant mappings that don't represent the true genomic structure.
- Adapter issues: Non-directional protocols may produce same-strand orientation. Preparing libraries with the NEXTFLEX™ Unique Dual Indexes, avoids this thanks to the use of directional adapter ligation. Additionally, incomplete adapter removal, incomplete trimming and index misassignment may complicate alignment.
Mitigating abnormal pair-end alignments
- Perform careful QC and adapter trimming: Always run FastQC or similar tools to identify quality drops or adapter contamination. Ensure trimming of adapter and low‑quality sequences before alignment.
- Use robust aligners and adjust settings: Employ aligners like BWA‑MEM or Bowtie2 with optimized settings, allowing for suboptimal hits to improve pairing accuracy. Some aligners (e.g., NovoAlign) perform well by considering multiple hits
- Filter chimeric reads: For FFPE or chimeric‑prone samples, apply specialized filtering (like MicroSEC) to remove artifacts from end‑repair microhomology.
- Validate insert size distributions: Monitor fragment size metrics; extreme variability may reflect library prep issues. Overlapping reads and short inserts should trigger adapter‑read‑through checks.
- Maintain read pairing order: Ensure FASTQ pairing is preserved across processing steps. Mis-ordered pairs can introduce false discordance Cross‑validate structural variant signals: For suspected structural variants, integrate paired‑end discordance with coverage anomalies and split‑read evidence. Use multiple callers (e.g., Delly, Manta) in combination to improve reliability, applying intersection‑union strategies
Conclusion
The challenge when discordant R1/R2 mapping appears lies in separating meaningful biological events such as structural variants from the noise. Careful QC, trimming, alignment parameter tuning, and multi-caller validation are essential steps to reduce artifacts and sharpen biological insights. Yet, the foundation of reliable paired-end data is set during library prep.
Using directional adapters and high-quality indexing strategies minimizes chimeras, adapter read-through, and misoriented pairs before they ever complicate downstream analysis. The NEXTFLEX™ Unique Dual Index Barcodes and our WGS
NEXTFLEX Rapid XP V2 DNA-Seq Kit
library preparation kits are specifically designed to deliver this robustness, ensuring that forward-reverse orientation is preserved and that true genomic signals stand out against the technical background.



